

Eukaryotic secretion pipeline
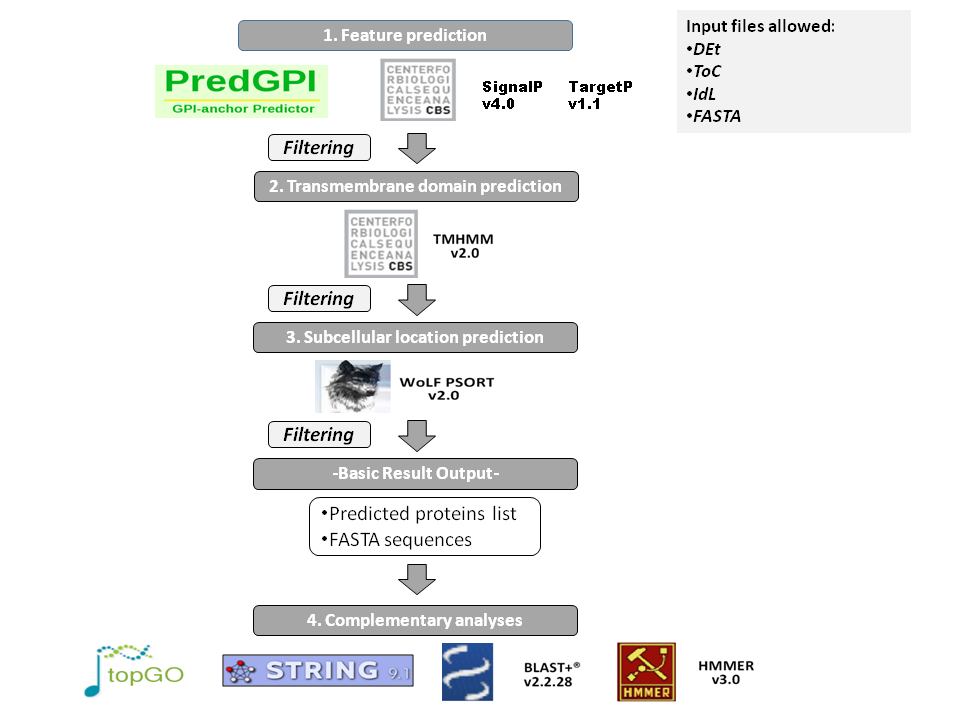
For a complete secretome analysis in eukaryotes, different tools for secretome analysis have been merged in a pipeline together with a set of Perl/Bioperl and R scripts. Tools for Eukaryotic analysis are presented under a one-step query interface, where users upload an input file in a format eligible between: Identifier lists (IDL) from different repositories, tables of counts (ToC), differential expresion tables (DEt) and FASTA files. Then selection of cut-off scores for the processing tools (or default values), and the optional tests to be performed (GO enrichment, Functional motifs and BLAST orthologous relations) can be chosen before data submission.
* Mandatory information